I治験と臨床研究、臨床試験
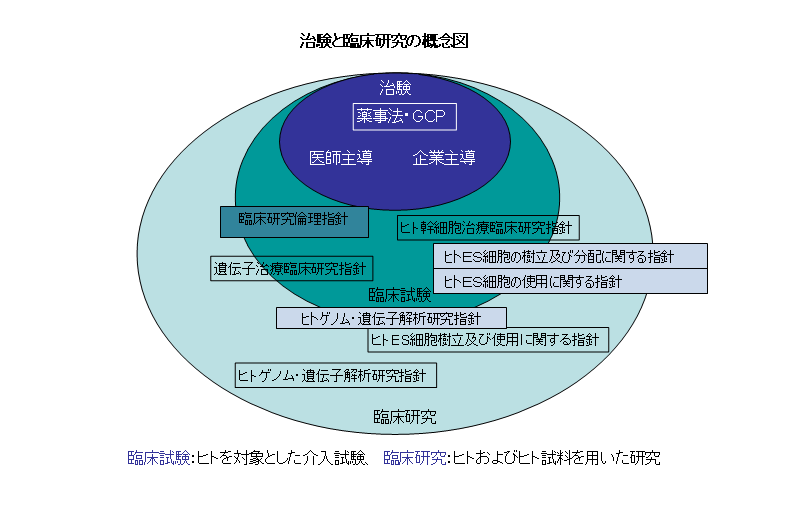
図-1.治験と臨床研究・臨床試験の概念図医薬品や医療機器を製品として販売や譲渡するには厚生労働大臣の製造販売承認を得る必要があります。製造販売承認申請(以下、「承認申請」)に必要な臨床試験の試験成績に関する資料の収集を目的とする試験は薬事法上「治験」と定義されています。治験では、非臨床試験で認められた効果や安全性をヒトで確認するため、科学的に評価できるデータとして倫理性・信頼性基準(GCP)のもとで収集することが求められています。GCPは、薬事法下の厚生労働省令で定められた基準であり、厳密な運用が行われています。治験以外のヒトを対象とした医師などが行う臨床研究については、厚生労働省が倫理指針を示しています(図-1)。
1.GCP(Good Clinical Practice)
治験を実施する場合、医薬品では「医薬品の臨床試験の実施の基準に関する省令」(H9.3.27 省令28)、医療機器では「医療機器の臨床試験の実施の基準に関する省令」(H17.3.23 省令36)を遵守しなければなりません。これらの省令は、GCP省令と呼ばれ、治験を倫理的かつ科学的に実施し、その信頼性を確保するための基準として制定され、国際的な整合性に配慮されています。GCP省令は治験を依頼する者、治験を自ら(医師主導治験)実施しようとする者に係る「治験の準備に関する基準」及び「治験の管理に関する基準」、治験を実施する医療機関が行うべき「治験を行う基準」などが定められています。最近の改正(医薬品についてはH20.2.29改正省令24、医療機器についてはH21.3.31改正省令68)では、治験を行うことの適否等を審議する治験審査委員会の審議の透明化(他施設の治験審査委員会への審査の依頼が可能)や情報公開(委員名簿や会議の記録の概要の公開)が定められました。
承認申請において提出された添付資料は、承認審査にあたり治験を依頼した企業や治験を実施した医療機関に対し、医薬品医療機器総合機構(総合機構)による信頼性調査としてGCP実地調査などの適合性調査が行われ、提出された資料が審査可能なデータであるか判断されます。
2.医師主導治験

図-2.平成15年改正薬事法における「医師主導の治験」
(厚生労働省のホームページより一部改変)平成15年以前の薬事法では、医療機関が治験を実施するのは企業が依頼した場合だけで、治験以外で企業が医療機関に未承認の薬物や未承認機器を提供することは認められていませんでした。また、医療現場でのニーズが高い医薬品や医療機器を医師などが研究開発した場合、臨床研究が実施されていながら再度企業が治験をやり直さなければならず、企業にとって採算が取れない製品の開発は進みませんでした。しかし、医薬品については平成15年の改正薬事法施行で、医療機器については平成17年の施行で、それぞれ医療機関・医師が自ら治験届を提出し、治験を実施することが可能となりました。これにより、医療機関が実施した先進的医療の臨床データを企業に引継ぐことができるようになり無駄な治験の繰り返しが省けることとなり、医療機関は薬事法上の承認がない薬物なども企業から提供を受けることが可能となりました。(図-2)
3.臨床研究倫理指針
治験以外の臨床研究については、「臨床研究に関する倫理指針」(H15.7.30告255)で、被験者の人権や安全確保のための倫理的な配慮が示されました。その後、国内の臨床試験の質の向上により、基礎の実績を迅速に臨床につなげ、さらには医薬品などとして実用化するために臨床研究倫理指針の改正(H20.7.31告415)が行われました。現行の指針では、倫理審査委員会の審議の透明化や情報公開、被験者の健康被害に対する補償措置や予期しない重篤な有害事象に対する対応、関係者の教育研修、および臨床試験計画の事前登録などが盛り込まれています。
なお、医薬品等の承認申請のために必要な臨床試験の資料はGCPに準拠して収集される必要があるため、臨床研究で収集された資料は、承認申請用の資料として利用することはできないので注意が必要です。