II新薬の研究開発・承認のプロセス
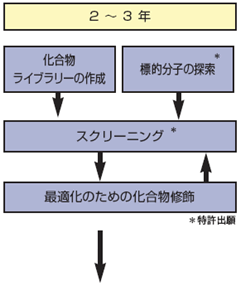
●化合物ライブラリーの作成
化合物を合成、培養、抽出などにより広範に収集し、数十万から数百万の化合物群からなるライブラリーを作成する。
●標的分子の探索
ゲノム、プロテオーム解析などを通じて、病態にかかわると考えられる標的分子を見つけだす。
●スクリ一ニング
まず、ライブラリーの中から、ハイスループット・スクリーニングなどの手法を用いて、新薬のもととなるリード化合物を見つけだす。さらに、リード化合物に化合物修飾を加えた化合物の中から、生化学、薬理、代謝、安全性研究などを通じて、薬効・安全性の両面から最適な化合物を選び出す。この段階をパスするのはごくわずかなものとなる。
●最適化のための化合物修飾
コンビナトリアルケミストリーなどの化学変換ほかにより、リード化合物の周辺化合物を数多く作る。
●特許出願
標的分子の探索時:標的分子の物質特許、機能に関する特許および標的分子を用いたスクリーニング法に関する特許。
スクリーニング時:新規物質の物質特許、既存物質の用途特許(新用途の発見に基づく)。
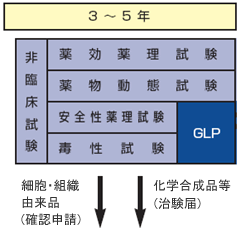
●薬効薬理試験
どれくらい与えると効果があるか、どのような方法で使用するかなどを調べる。
●薬物動態試験
体内でどのように吸収され、分布し、排泄されるかなどを調べる。
●安全性薬理試験
大量投与されたときなどに主な生理機能に対して医薬品としてどのような望ましくない作用があるのかを調べる。
●毒性試験
一般的な医薬品ではげっ歯類やイヌ、サルで急性、亜急性、慢性毒性試験を実施する。また医薬品の特性に応じて、がん原性や依存性などの毒性試験が必要となる。
こうしたことを徹底的に調べ、ヒトに対する安全性を予測したうえで臨床試験に移る。
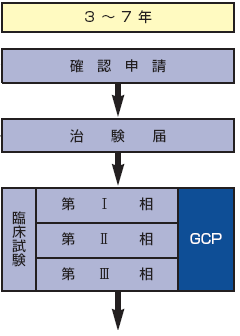
●治験
新薬の承認申請に必要なデータを作成するためにヒトを対象に行う試験。被験者に試験の目的や内容を十分に説明し、文書による同意を得ることが求められている(インフォームド・コンセント)。
●第I相(Phase I)
同意を得た少数の健康人志願者を対象に、安全性のテストを行う。
●第II相(Phase II)
同意を得た少数の患者を対象に有効で安全な投薬量や投薬方法などを確認する。
●第III相(Phase III)
同意を得た多数の患者で、「二重盲検試験」などにより、既存薬などと比較して新薬の有効性および安全性をチェックする。
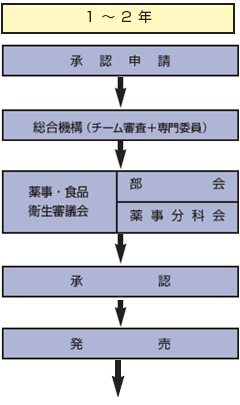
●承認申請
医療上の有効性と安全性が確認された新薬について、製薬企業は厚生労働省に製造販売承認の申請を行う。これを受けて厚生労働省は総合機構における審査にかけ、その結果をもって、厚生労働大臣の諮問機関である薬事・食品衛生審議会に諮る。審査をパスしたものには、厚生労働大臣から製造販売承認が与えられる。
●審査
承認申請資料の審査は、医学、薬学、生物統計学などの分野別の専門官によるチーム審査が行われ、さらに臨床家などの立場からの専門委員の意見を踏まえ審査報告書が作成される。
●薬価基準収載申請
製薬企業は製造販売承認が与えられた新薬について、薬価基準収載の申請を行う。
●製剤開発のプロセス
 新薬のモトとなる物質の物理的・化学的性状およびより合理的な製造方法などを調べる。
新薬のモトとなる物質の物理的・化学的性状およびより合理的な製造方法などを調べる。●製剤開発
新薬の実生産に向けて、製品の規格や試験方法などを設定するとともに、GMPに適合した治験薬を供給する。